Un nuovo approccio alla fibrosi cistica

Ph: medicalnewstoday.com
Chi ha conosciuto da vicino la fibrosi cistica sa quanto può essere complessa. Si deve affrontare il fatto che cure risolutive non ce ne sono ma pian piano la ricerca è riuscita a prolungare la vita di questi pazienti. Fino a qualche decennio fa si diceva: “Guai a quel bambino che, se baciato sulla fronte, sa di sale”. Il sudore molto salato era un segno tipico dei bambini affetti da fibrosi cistica e per loro si poteva fare ben poco.
Come riporta l’Osservatorio Malattie Rare (OMAR), nonostante l’incidenza della patologia sia di 1 su 2500 in Italia non è considerata come una patologia rara. È stata oggetto di una legge ad hoc che, nel 1993, la definì come “di alto interesse sociale” invitando le Regioni ad intraprendere un programma di diagnosi precoce.
La nuova sfida americana
Pubblicato sulla rivista Advanced Materials (disponibile qui), il nuovo studio dei ricercatori del MIT apre nuove strade per provare a contrastare la fibrosi cistica.
La causa di questa malattia è la presenza di mutazioni all’interno del cromosoma 7. Queste non permettono la corretta produzione di una proteina, CFTR, che è un canale del cloro. Tale mancanza comporta la produzione di un muco molto denso e molto viscoso da parte da pare di quelle ghiandole che riversano il proprio secreto all’esterno dell’organismo.
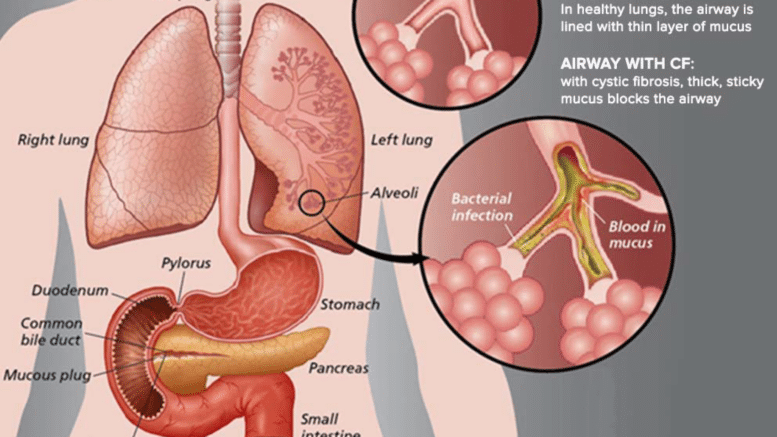
Le problematiche polmonari sono le più importanti dal punto di vista della mortalità legata alla malattia. Espellere un muco così denso dall’albero bronchiale, infatti, è molto difficoltoso e il suo accumulo comporta la creazione di un ambiente particolarmente adatto allo sviluppo batterico. Questi pazienti, infatti, si trovano spesso a fare i conti con infezioni polmonari molto severe.
Le nuove scoperte in ambito di ingegneria genetica, tuttavia, iniziano a mostrare la loro utilità anche in questo campo. Se il problema è che non viene trascritto il gene CFTR nulla vieta che la cellula sia comunque in grado di produrre la proteina, almeno in teoria. In fondo anche l’essere umano funziona così: leggere una ricetta per preparare una torta significa seguire delle istruzioni.
Ma una cellula? Come possiamo dirgli di produrre qualcosa che non è scritto nel DNA?
L’inalazione delle informazioni
La metodica sviluppata dai ricercatori del MIT in collaborazione con Translate Bio mira proprio a risolvere questo problema. Se si riuscisse a portare un RNA messaggero prodotto in laboratorio all’interno delle cellule, queste potrebbero tradurlo in proteine.
I ricercatori sono riusciti in questo intento. Creando un polimero carrier in grado di ingannare le cellule polmonari sono stati in grado di far produrre l’enzima luciferasi, ovvero l’enzima che permette alle lucciole di emanare luce, a delle cellule di polmone di topo, sia in vitro che in vivo. Per far ciò hanno nebulizzato un mRNA legato al carrier e lo hanno fatto inalare alla cavia. La cosa molto interessante è che il farmaco non veniva riscontrato in nessun altro tessuto al di fuori di quello polmonare, il che consente di limitare gli effetti collaterali indesiderati.
L’idea è che al posto della codifica della luciferasi possa essere inserita quella per CFTR funzionante. In questo modo si potrebbe quanto meno alleviare la sintomatologia della fibrosi cistica, migliorando la qualità e la durata della vita dei pazienti.
Al momento sono in corso i trials di fase 1 e 2 per valutare la reale efficacia di questo approccio. La speranza è quella di avere, quanto prima, una soluzione importante per una patologia difficile come la fibrosi cistica.
Fonte:
Advanced Materials: Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium